3. Generating a 4D-Communication Tensor from computed communication scores
After inferring communication scores for combinations of ligand-receptor and sender-receiver cell pairs, we can use that information to identify context-dependent CCC patterns across multiple samples simultaneously by generating a 4D-Communication Tensor. LIANA handily outputs these score as a dataframe that is easy to use for building our tensor.
In this tutorial we will show you how to use the dataframe saved from LIANA to generate a 4D-Communication Tensor that could be later used with Tensor-cell2cell.
Initial Setup
Import the necessary packages
[1]:
import cell2cell as c2c
import liana as li
import numpy as np
import pandas as pd
Directories
[2]:
data_folder = '../../data/liana-outputs/'
[3]:
output_folder = '../../data/tc2c-outputs/'
c2c.io.directories.create_directory(output_folder)
../../data/tc2c-outputs/ already exists.
Load Data
Open the dataframe containing LIANA results for every sample/context (this can be also found in adata.uns['liana_res']. These results contain the communication scores of the combinations of ligand-receptor pairs and sender-receiver pairs.
[4]:
liana_res = pd.read_csv(data_folder + 'LIANA_by_sample.csv')
Create 4D-Communication Tensor
Specify the order of the samples/contexts
Here, we will specify an order of the samples/contexts given the condition they belong to (HC or Control, M or Moderate COVID-19, S or Severe COVID-19).
[5]:
sorted_samples = sorted(liana_res['sample_new'].unique())
[6]:
sorted_samples
[6]:
['HC1', 'HC2', 'HC3', 'M1', 'M2', 'M3', 'S1', 'S2', 'S3', 'S4', 'S5', 'S6']
[7]:
liana_res.head()
[7]:
| sample_new | source | target | ligand_complex | receptor_complex | lr_means | cellphone_pvals | expr_prod | scaled_weight | lr_logfc | spec_weight | lrscore | lr_probs | cellchat_pvals | specificity_rank | magnitude_rank | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | HC1 | Macrophages | NK | B2M | CD3D | 3.410504 | 0.0 | 8.059611 | 1.300556 | 1.397895 | 0.083273 | 0.961040 | 0.221495 | 0.0 | 0.003713 | 1.698996e-09 |
| 1 | HC1 | T | NK | B2M | CD3D | 3.410586 | 0.0 | 8.059861 | 1.300856 | 1.272266 | 0.083276 | 0.961041 | 0.221213 | 0.0 | 0.003713 | 6.256593e-09 |
| 2 | HC1 | NK | NK | B2M | CD3D | 3.264099 | 0.0 | 7.614378 | 0.790913 | 1.113901 | 0.078673 | 0.959963 | 0.216816 | 0.0 | 0.006245 | 2.653267e-08 |
| 3 | HC1 | T | NK | B2M | KLRD1 | 3.297900 | 0.0 | 6.865250 | 6.960920 | 1.244892 | 0.171293 | 0.957924 | 0.214586 | 0.0 | 0.000092 | 9.767878e-08 |
| 4 | HC1 | Macrophages | NK | B2M | KLRD1 | 3.297818 | 0.0 | 6.865037 | 6.960620 | 1.370520 | 0.171288 | 0.957924 | 0.214861 | 0.0 | 0.000092 | 1.086199e-07 |
Generate tensor
To generate the 4D-communication tensor, we will to create matrices with the communication scores for each of the ligand-receptor pairs within the same sample, then generate a 3D tensor for each sample, and finally concatenate them to form the 4D tensor.
Briefly, we use the LIANA dataframe and communication scores to organize them as follows:
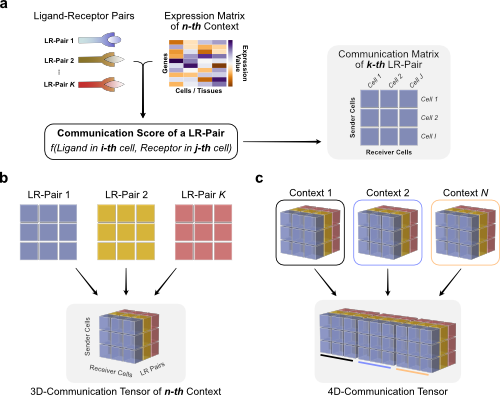
LIANA includes a function that does all these steps at once.
We will transform the structure of the communication scores from a set of 2D-matrices for each sample into a 3D Tensor where the third dimension is sample/context.
The key parameters when building a tensor are:
non_negativeandnon_negative_fillas Tensor-cell2cell by default expects non-negative values, any values below 0 will be set to 0 (e.g. this is relevant if one wants to use e.g. theLRlog2FCscore function). If you used a pipeline that generated negative scores, we suggest replacing these with 0. Otherwise, by default, Tensor-cell2cell will treat these as NaN. Since we used the magnitude rank score, which is non-negative, these parameters won’t affect our results.inverse_funis the function we use to convert the communication score before using it to build the tensor. In this case, the ‘magnitude_rank’ score generated by LIANA considers low values as the most important ones, ranging from 0 to 1. In contrast, Tensor-cell2cell requires higher values to be the most important scores, so here we pass a function (lambda x: 1 - x) to adapt LIANA’s magnitude-rank scores (subtracts the LIANA’s score from 1). IfNoneis passed instead, no transformation will be performed on the communication score. If using other scores coming from one of the methods implemented in LIANA, a similar transformation can be done depending on the parameters and assumptions of the scoring method.non_expressed_fillindicates what value to assign to missing scores when liana was run withreturn_all_lrsis set toTrue(i.e., those that did not passed LIANA’s filters and were not inferred because ligands and/or receptors were not expressed; see parameterexpr_propin the Notebook for Inferring the Communication Scores). IfNoneis passed, missing values will be treated asnumpy.nanvalues. In this example, this is not used as we use theouter_fractionparameter from tensor (see below) to address this.howcontrols which ligand-receptor pairs and cell types to include when building the tensor. This decision depends on whether the missing values across a number of samples for both ligand-receptor interactions and sender-receiver cell pairs are considered to be biologically-relevant. Options are:'inner'is the more strict option since it only considers only cell types and LR pairs that are present in all contexts (intersection).'outer'considers all cell types and LR pairs that are present across contexts (union).'outer_lrs'considers only cell types that are present in all contexts (intersection), while all LR pairs that are present across contexts (union).'outer_cells'considers only LR pairs that are present in all contexts (intersection), while all cell types that are present across contexts (union).
The following two parameters (``lr_fill`` and ``cell_fill``) indicate what value to assign missing scores when ``how`` is not set to ``’inner’``, i.e., there are cells or LR pairs that are not present in all contexts. During tensor component analysis, NaN values are masked such that they are not considered by the decomposition objective. This results in behavior of NaNs being imputed as missing values that are potentially communicating, whereas if missing LRs are filled with a value such as 0, they are treated as biological zeroes (i.e., not communicating). For additional details and discussion regarding this parameter, please see the missing indices benchmarking.
lr_fillis the value to fill communication scores when a ligand-receptor pair is not use by any cell type within a sample. Here we treat these cases as missing values by passing anumpy.nanvalue.cell_fillis the value to fill communication scores when a cell type is not using a given ligand-receptor pair within a sample. This value has priority overlr_fillif that ligand-receptor pair is used at least by one pair of the sender-receiver cell pairs within the sample. Here we treat these cases as missing values by passing anumpy.nanvalue.outer_fractioncontrols the elements to include in the union scenario of thehowoptions. Only elements that are present at least in this fraction of samples/contexts will be included. When this value is 0, the tensor includes all elements across the samples. When this value is 1, it acts as usinghow='inner'.
In this case we will consider cell types and LR pairs that are in the LIANA results at least in 1/3 of the samples
[8]:
tensor = li.multi.to_tensor_c2c(liana_res=liana_res, # LIANA's dataframe containing results
sample_key='sample_new', # Column name of the samples
source_key='source', # Column name of the sender cells
target_key='target', # Column name of the receiver cells
ligand_key='ligand_complex', # Column name of the ligands
receptor_key='receptor_complex', # Column name of the receptors
score_key='magnitude_rank', # Column name of the communication scores to use
non_negative = True, # set negative values to 0
inverse_fun=lambda x: 1 - x, # Transformation function
non_expressed_fill=None, # Value to replace missing values with
how='outer', # What to include across all samples
lr_fill=np.nan, # What to fill missing LRs with
cell_fill = np.nan, # What to fill missing cell types with
outer_fraction=1/3., # Fraction of samples as threshold to include cells and LR pairs.
lr_sep='^', # How to separate ligand and receptor names to name LR pair
context_order=sorted_samples, # Order to store the contexts in the tensor
sort_elements=True # Whether sorting alphabetically element names of each tensor dim. Does not apply for context order if context_order is passed.
)
100%|███████████████████████████████████████████| 12/12 [00:18<00:00, 1.53s/it]
Evaluate some tensor properties
Tensor shape
This indicates the number of elements in each tensor dimension: (Contexts, LR pairs, Sender cells, Receiver cells)
[9]:
tensor.shape
[9]:
(12, 1054, 10, 10)
Missing values
This represents the fraction of values that are missing. In this case, missing values are combinations of contexts x LR pairs x Sender cells x Receiver cells that did not have a communication score or were missing in the dataframes.
[10]:
tensor.missing_fraction()
[10]:
0.906289531941809
Sparsity
This represents the fraction of values that are a real zero (excluding the missing values)
[11]:
tensor.sparsity_fraction()
[11]:
0.04997707147375079
Fraction of excluded elements
This represents the fraction of values that are ignored (masked) in the analysis. In this case it coincides with the missing values because we did not generate a new tensor.mask to manually ignore specific values. Instead, it automatically excluded the missing values.
[12]:
tensor.excluded_value_fraction() # Percentage of values in the tensor that are masked/missing
[12]:
0.906289531941809
Prepare Tensor Metadata
To interpret analysis on the tensor, we can assign groups to each sample/context, and to every elements in the other dimensions (LR pairs and cells).
We can generate respective dictionaries manually or automatically from DBs.
Default dict to return Unknown if major groups are not present for a given element
[13]:
from collections import defaultdict
element_dict = defaultdict(lambda: 'Unknown')
Major groups of the samples/contexts
Please note that this context_dict could be directly generated from the adata object in the Notebook for Inferring the Communication Scores by using the command:
context_dict = adata.obs.set_index('sample_new')['condition'].sort_values().to_dict()
[14]:
context_dict = element_dict.copy()
context_dict.update({'HC1' : 'Control',
'HC2' : 'Control',
'HC3' : 'Control',
'M1' : 'Moderate COVID-19',
'M2' : 'Moderate COVID-19',
'M3' : 'Moderate COVID-19',
'S1' : 'Severe COVID-19',
'S2' : 'Severe COVID-19',
'S3' : 'Severe COVID-19',
'S4' : 'Severe COVID-19',
'S5' : 'Severe COVID-19',
'S6' : 'Severe COVID-19',
})
dimensions_dict = [context_dict, None, None, None]
Generate a list containing metadata for each tensor order/dimension - Later used for coloring factor plots
[15]:
meta_tensor = c2c.tensor.generate_tensor_metadata(interaction_tensor=tensor,
metadata_dicts=[context_dict, None, None, None],
fill_with_order_elements=True
)
If you want to color the elements of another dimension by major groups, just replace the corresponding None in metadata_dicts=[context_dict, None, None, None] by a dictionary whose keys are the element names of the dimension and the values are the major groups. For example, if you want to color LR pairs, you should create a dictionary whose keys are the names from tensor.order_names[1], and put that new dictionary (e.g. lr_dict) in
metadata_dicts=[context_dict, lr_dict, None, None]. For sender and receiver cells, the same could be done.
Export Tensor
Here we will save the tensor as a pickle object with cell2cell, so we can use it later with other analyses.
[16]:
c2c.io.export_variable_with_pickle(tensor, output_folder + '/BALF-Tensor.pkl')
../../data/tc2c-outputs//BALF-Tensor.pkl was correctly saved.
Export Tensor Metadata
[17]:
c2c.io.export_variable_with_pickle(meta_tensor, output_folder + '/BALF-Tensor-Metadata.pkl')
../../data/tc2c-outputs//BALF-Tensor-Metadata.pkl was correctly saved.
Make sure to use this pandas version to load the metadata in the future to avoid errors
[18]:
pd.__version__
[18]:
'2.1.4'
Supplementary Information about Tensor-cell2cell
The function li.multi.to_tensor_c2c() from LIANA that we used to build the tensor relies on the function c2c.tensor.dataframes_to_tensor() from cell2cell. We can use the cell2cell’s function instead for more fine parameter tuning, as follows:
First, we need to create a dictionary with sample names as keys and dataframes containing the communication scores within each sample. Here we split the LIANA output to recreate that.
[19]:
data = dict(list(liana_res.groupby('sample_new')))
This is, for example, the dataframe for the sample HC1
[20]:
data['HC1']
[20]:
| sample_new | source | target | ligand_complex | receptor_complex | lr_means | cellphone_pvals | expr_prod | scaled_weight | lr_logfc | spec_weight | lrscore | lr_probs | cellchat_pvals | specificity_rank | magnitude_rank | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | HC1 | Macrophages | NK | B2M | CD3D | 3.410504 | 0.0 | 8.059611 | 1.300556 | 1.397895 | 0.083273 | 0.961040 | 0.221495 | 0.0 | 0.003713 | 1.698996e-09 |
| 1 | HC1 | T | NK | B2M | CD3D | 3.410586 | 0.0 | 8.059861 | 1.300856 | 1.272266 | 0.083276 | 0.961041 | 0.221213 | 0.0 | 0.003713 | 6.256593e-09 |
| 2 | HC1 | NK | NK | B2M | CD3D | 3.264099 | 0.0 | 7.614378 | 0.790913 | 1.113901 | 0.078673 | 0.959963 | 0.216816 | 0.0 | 0.006245 | 2.653267e-08 |
| 3 | HC1 | T | NK | B2M | KLRD1 | 3.297900 | 0.0 | 6.865250 | 6.960920 | 1.244892 | 0.171293 | 0.957924 | 0.214586 | 0.0 | 0.000092 | 9.767878e-08 |
| 4 | HC1 | Macrophages | NK | B2M | KLRD1 | 3.297818 | 0.0 | 6.865037 | 6.960620 | 1.370520 | 0.171288 | 0.957924 | 0.214861 | 0.0 | 0.000092 | 1.086199e-07 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 4218 | HC1 | T | T | LGALS3BP | CD33 | 0.546831 | 1.0 | 0.162326 | -0.428778 | -0.330954 | 0.065941 | 0.777816 | 0.000000 | 1.0 | 1.000000 | 1.000000e+00 |
| 4219 | HC1 | T | T | C1QB | CD33 | 1.500021 | 1.0 | 0.499953 | -0.674297 | -0.418023 | 0.050734 | 0.860018 | 0.000000 | 1.0 | 1.000000 | 1.000000e+00 |
| 4220 | HC1 | T | T | C1QA | CD33 | 1.491162 | 1.0 | 0.496815 | -0.682417 | -0.439526 | 0.058043 | 0.859639 | 0.000000 | 1.0 | 1.000000 | 1.000000e+00 |
| 4221 | HC1 | T | T | LGALS1 | CD69 | 1.350314 | 1.0 | 0.522723 | -0.108787 | 0.406618 | 0.039087 | 0.862677 | 0.000000 | 1.0 | 1.000000 | 1.000000e+00 |
| 4222 | HC1 | mDC | mDC | CIRBP | TREM1 | 0.634905 | 1.0 | 0.233897 | -0.544721 | -0.515300 | 0.013720 | 0.807776 | 0.000000 | 1.0 | 1.000000 | 1.000000e+00 |
4223 rows × 16 columns
We can check what samples are included
[21]:
data.keys()
[21]:
dict_keys(['HC1', 'HC2', 'HC3', 'M1', 'M2', 'M3', 'S1', 'S2', 'S3', 'S4', 'S5', 'S6'])
As explained before, the magnitude_rank score needs to be converted before using it with Tensor-cell2cell. Thus, we modify it here for each of the sample dataframes.
[22]:
for sample, df in data.items():
df['magnitude_rank'] = df['magnitude_rank'].apply(lambda x: 1-x)
data[sample] = df
The key parameters when building a tensor with the cell2cell’s function are:
howcontrols what ligand-receptor pairs and cell types to include when building the tensor. This decision is dependent on the number of samples including scores for their combinations of ligand-receptor and sender-receiver cell pairs. Options are:'inner'is the more strict option since it only considers only cell types and LR pairs that are present in all contexts (intersection).'outer'considers all cell types and LR pairs that are present across contexts (union).'outer_lrs'considers only cell types that are present in all contexts (intersection), while all LR pairs that are present across contexts (union).'outer_cells'considers only LR pairs that are present in all contexts (intersection), while all cell types that are present across contexts (union).
The following two parameters (``lr_fill`` and ``cell_fill``) indicate what value to assign missing scores when ``how`` is not set to ``’inner’``, i.e., there are cells or LR pairs that are not present in all contexts. During tensor component analysis, NaN values are masked such that they are not considered by the decomposition objective. This results in behavior of NaNs being imputed as missing values that are potentially communicating, whereas if missing LRs are filled with a value such as 0, they are treated as biological zeroes (i.e., not communicating). For additional details and discussion regarding this parameter, please see the missing indices benchmarking.
lr_fillis the value to fill communication scores when a ligand-receptor pair is not use by any cell type within a sample. Here we treat these cases as missing values by passing anumpy.nanvalue.cell_fillis the value to fill communication scores when a cell type is not using a given ligand-receptor pair within a sample. This value has priority overlr_fillif that ligand-receptor pair is used at least by one pair of the sender-receiver cell pairs within the sample. Here we treat these cases as missing values by passing anumpy.nanvalue.outer_fractioncontrols the elements to include in the union scenario of thehowoptions. Only elements that are present at least in this fraction of samples/contexts will be included. When this value is 0, considers all elements across the samples. When this value is 1, it acts as usinghow='inner'.
In this case we will consider cell types and LR pairs that are in the LIANA results at least in 1/3 of the samples
[23]:
tensor = c2c.tensor.dataframes_to_tensor(context_df_dict=data,
sender_col='source', # Column name of the sender cells
receiver_col='target', # Column name of the receiver cells
ligand_col='ligand_complex', # Column name of the ligands
receptor_col='receptor_complex', # Column name of the receptors
score_col='magnitude_rank', # Column name of the communication scores
how='outer', # What to include across all samples
outer_fraction=1/3., # Fraction of samples as threshold to include cells and LR pairs.
lr_sep='^', # How to separate ligand and receptor names to name LR pair
context_order=sorted_samples, # Order to store the contexts in the tensor
sort_elements=True, # Whether sorting alphabetically element names of each tensor dim. Does not apply for context order if context_order is passed.
lr_fill=np.nan,
cell_fill=np.nan,
)
100%|███████████████████████████████████████████| 12/12 [00:18<00:00, 1.53s/it]
[24]:
tensor.shape
[24]:
(12, 1054, 10, 10)